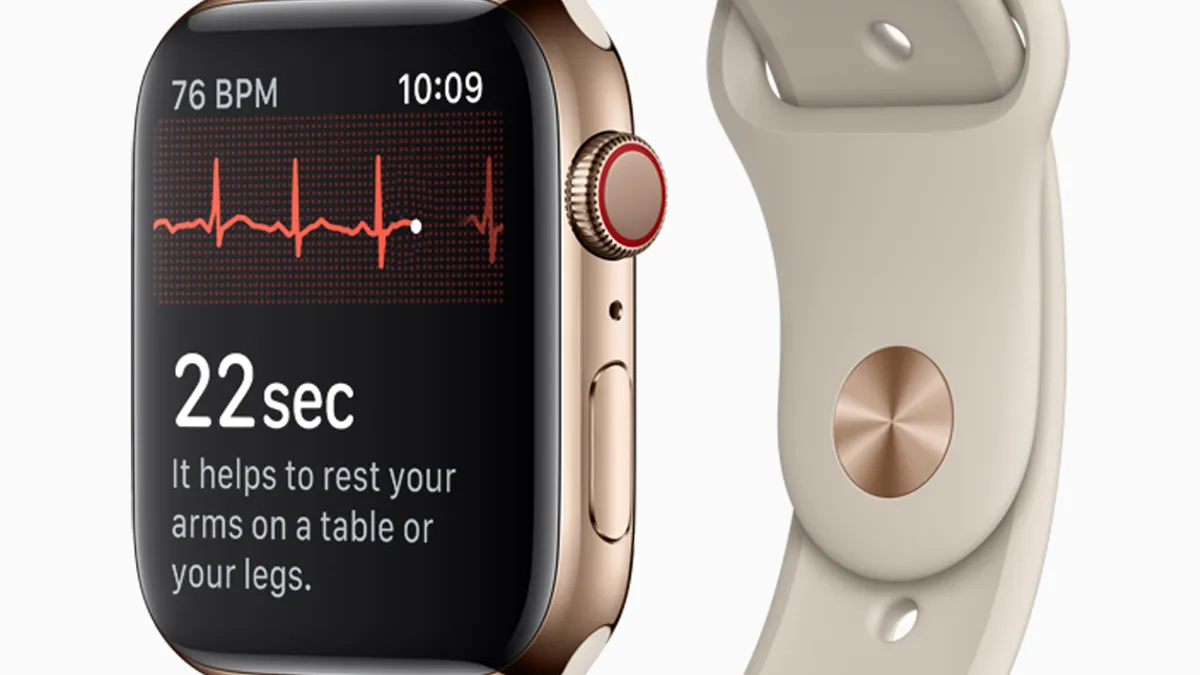
The Apple Watch's electrocardiogram capability, which rolled out last week, was among the high-profile FDA device clearances of 2018, just one example of Silicon Valley putting its stamp on the medical device landscape this year.
Digital health companies contributed to the 38 De Novo authorizations the agency has granted so far this year, which already tops the 31 clearances in all of 2017. But neither the 510(k) nor premarket approval (PMA) pathways have shown a similar uptick shortly before the end of the year, according to publicly available data.
The number of De Novo OKs — including Apple's EKG — has grown as the agency seeks to further encourage use of the pathway, ultimately seeking a wave of new predicates for 510(k)s as it proposes updates to its De Novo process. The De Novo makeover goes hand in hand with FDA's effort to reduce 510(k) application reliance on predicates older than 10 years.
The decades-old 510(k) program, which the vast majority of products currently use, has come under fire in recent months for speeding too many devices through with limited regulatory scrutiny.
With two weeks to go before the year ends, MedTech Dive rounded up other notable 2018 approval actions:
Tech firms looking for FDA approval
The year saw the first FDA-approved direct-to-consumer birth control app and a downloadable Apple Watch feature that can detect irregular heart rhythms that may portend atrial fibrillation, along with the smart watch's electrocardiogram.
All three of these — joining 35 other De Novo authorizations thus far this year — were examples of the traditionally less-trafficked pathway at work.
The rise in digital product clearances underscores tech companies' increasing desire to shift the classification of fitness trackers and smartphone apps from entertainment to clinically-vetted medical devices with FDA imprimatur, says Allyson Hein, medical device lead at life sciences and consumer products firm Clarkston Consulting. And it's impacting how other device companies look at their product pipelines.
"The digital push from high-tech leaders is pushing traditional medical device manufacturers into new territory with faster product development timelines to meet consumers' growing demand for technology and personalization," Hein told MedTech Dive via email.
Next-generation competencies like advanced machine learning and the use of artificial intelligence in care decisions are already impacting device development.
"Devices focused on diagnostics are using machine learning algorithms to supplement and eventually replace requirements for clinician review in diagnosis," she said. "This also drives blended portfolio lines for pharma and biotech companies as they recognize the value of combination and companion products along with supporting software applications — which are classified as devices."
50 PMAs get FDA nod
So far this year, there have been 28 original and 22 panel track supplements, for a total of 50 PMA approvals, slightly down from a combined 64 at the close of 2017 (46 originals and 18 supplements.) Five of the approvals this year were for glucose monitoring systems, including Abbott's FreeStyle Libre blood sugar sensor for 14-day use. The system's two-week capacity makes it the longest-lasting self-applied personal glucose monitoring system on the market and is four days longer than Abbott's 10-day sensor system, which was approved in 2017.
FDA also approved the first artificial iris. Human Optics' Custom Flex artificial iris is designed for use in patients with no iris or eye damage.
Other approvals include Medtronic's DBS epilepsy system, approved as an add-on therapy for uncontrolled focal epilepsy, and Abbott's HeartMate 3 left ventricular assist system, whose approval was expanded to include destination therapy for patients with advanced heart failure. That means HeartMate 3 can now be used as a long-term implant in patients not eligible for a heart transplant.
The batch of approvals also includes a combination product and a companion diagnostic product. FDA approved Wright Medical's Augment Injectable combo bone graft material for use in hindfoot and ankle fusions.
And it greenlighted Foundation Medicine's FoundationFocus CDx. The assay detects BRCA1 and BRCA2 genes to determine if patients previously treated for ovarian cancer could benefit from Rubraca (rucaparib). That drug is a first-in-class anticancer agent targeting the DNA repair enzyme poly-ADP ribose polymerase (PARP).
This year's PMA approvals also included one expedited review, for TransMedics' Organ Care System Lung System, a normothermic machine perfusion system for preserving standard criteria donor lungs prior to transplantation.
Renewed focus on 510(k)s
Still, the majority of new products OK'd for marketing get there via the 510(k) route. As of Dec. 10, 2,888 510(k) authorizations had been granted in 2018, according to FDA. In 2017, the number of 510(k)s for the entire year totaled 3,173, representing 97% of total devices cleared or approved that year.
Among recent clearances are Viseon's spinal surgery device, which captures images of spinal surgery targets for display on operating room monitors, and RightEye's eye-tracking system, a device to detect and analyze vision impairment.
Hein predicts a renewed focus on 510(k)s in the coming year, as FDA seeks to process a wave of newer technologies, many of them digital or AI-based.
"The FDA will push for companies to use comparisons to more recently approved devices when seeking [clearance] through the 510(k) process," she said. "Current proposals do not call for any increase in clinical testing to win [clearance] through this pathway, which is important for an industry where changes typically occur incrementally."
Industry will also see a continued opportunity for IoT-connected smart devices to provide devicemakers with a wide range of new data sets with potential applications across their businesses, Hein said. "The proactive application and use of this data can lead to a better consumer experience in an increasingly patient-centric industry," she added.